
Contents
Group of disorders affecting the renal tubules characterized by an impaired ability to acidify the urine and excrete acid.
Mnemonic device:
Physiology
Normal Acid–Base Homeostasis:
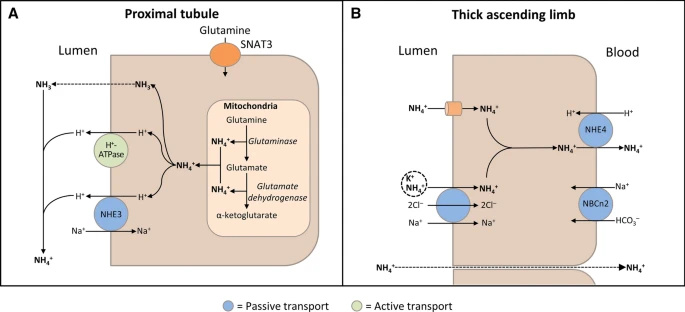
The kidneys contribute to the maintenance of acid–base homeostasis by reabsorbing HCO3− in the proximal tubule, as well as regenerating HCO3– in the cortical collecting duct

| Palmer BF. Normal acid-base balance. In: Johnson RJ, Feehally J, Floege J, editors. Comprehensive clinical nephrology. 5th ed. Philadelphia: Elsevier; 2014. p. 142–8.
Etiopathogeneis
Classification:
- Type 1: Distal RTA
- Type 2: Proximal RTA
- Type 3: Mixed RTA
- Type 4: Hyporeninemic hypoaldosteronism RTA

| Palmer, B.F., Kelepouris, E. & Clegg, D.J. Renal Tubular Acidosis and Management Strategies: A Narrative Review. Adv Ther 38, 949–968 (2021). https://doi.org/10.1007/s12325-020-01587-5
Type 1 (distal RTA) “Classical RTA”:
Distal tubule is responsible for generating new bicarbonate under influence of aldosterone. Damage to α-intercalated cells of distal tubule causes no new generation of HCO3– and thus no H+. This raises the pH of urine due to an inability to excrete acid and generate acidic urine in the distal tubule, even in states of metabolic acidosis.
Defective H+ ion secretion by α-intercalated cells in late DCT & CT → ↑ H+ ions (blood acidemia)
- Autoimmune diseases: Sjogren’s syndrome, SLE
- Liver cirrhosis
- Medullary sponge kidney
- Drugs: Amphotericin B, lithium)
Type 2 (proximal) RTA:
Normally 85% to 90% of bicarbonate is reabsorbed at proximal tubule, and only 10% reabsorbed at distal tubule. Due to a HCO3– leak, impaired proximal HCO3– reabsorption in proximal tubule results in excess HCO3–in urine leading to metabolic acidosis.
Impaired HCO3– reabsorption in PCT → ↑ HCO3– loss in urine → ↑ Blood acidity
- Multiple myeloma
- Fanconi syndrome
- Lowe syndrome
- Dent’s disease
- Amyloidosis
- Metabolic disease: Wilson disease, hereditary fructose intolerance, cystinosis, galactosemia
- Drugs: Acetazolamide, outdated tetracycline
Type 3 (mixed) RTA:
Rare inherited type 3 RTA is caused by mutations of CA II (chromosome 8q22) resulting in carbonic anhydrase II deficiency.
- Kartagener’s syndrome (KS)
Type 4 (hyperkalemic) RTA: Hyporeninemic hypoaldosteronism RTA
Hypoaldosteronism causes hyperkalemia and metabolic acidosis. Hyperkalemia impairs ammonia genesis in the proximal tubule and reduces the availability of NH3 to buffer urinary H+ and decreases H+ excretion in urine. The ability to acidify urine in this type of RTA is due to the inadequate amount rather than the complete absence of NH3 available for buffering of protons. Even if only a few protons are secreted distally, urine pH will fall, and this is why these patients have a urine pH < 5.5.
Aldosterone deficiency/resistance in CT → ↑ K+ & ↓ Na+
M/C type and always acquired
- Diabetic nephropathy (M/C cause)
- Addison’s disease
- SLE
- Sickle cell disease
- Obstructive uropathy
- Drugs: Trimethoprim, NSAIDs, ACE inhibitors, spironolactone)

Presentation
General symptoms:
D/t metaoblic acidosis
- Gastrointestinal features: ↓ apetite, vomiting, abdominal pain
- Kussmaul breathing (hyperventilation in response to acidosis): Rapid, shallow breaths that get deeper over time (to blow off CO2)

Distal RTA (type 1 RTA):
Commonly observed in children as a primary genetic defect. Acquired deficits usually manifest in adults and are most typically associated with autoimmune conditions or nephrotoxins.
- Vomiting and profound dehydration
- Obtundation (reduced level of alertness/consciousnes)
- Restricted skeletal growth
- Rickets
Proximal RTA (type 2 RTA):
Most cases are discovered in children with heritable mutations
- Mild disease: Short stature and lethargy
- Severe cases: Respiratory distress, vomiting, and feeding difficulties
Fanconi syndrome:
Fanconi syndrome is a malabsorptive state of the proximal convoluted tubules, while proximal RTA refers only to the deficiency in HCO3− retention
Most cases of proximal RTA occur as a component of Fanconi syndrome
- Failure to thrive
- Volume depletion
- Rickets
- Constipation
- Rare genetic disorders associated with Fanconi syndrome can manifest:
- Ocular abnormalities
- Tooth deformities
- Intellectual impairment
Hyperkalemic RTA (type 4 RTA):
Manifestations are a consequence of the effects of aldosterone deficiency or insensitivity. Biochemical findings are routinely the only detectable change in these patients.
- Postural hypotension and lethargy
Mixed syndrome (type 3 RTA):
Rarely seen in clinical practice and has diagnostic findings characteristic of both distal and proximal RTA
- Debilitating congenital syndromes in children and is principally associated with carbonic anhydrase II deficiency
Long term complications:
- Shock (d/t vasodilation of peripheral arterioles)
- Nephrolithiasis (calcium phosphate stones) is frequently associated with untreated type 1 RTA.

Diagnosis
In general, RTA should be suspected when metabolic acidosis is accompanied by hyperchloremia and a normal plasma anion gap (Na+ − [Cl− + HCO3−] = 8 to 16 mmol/L) in a patient without evidence of gastrointestinal HCO3− losses and who is not taking acetazolamide or ingesting exogenous acid.
Biochemical findings:
In all cases of metabolic acidosis, plasma or serum anion gap should be the first laboratory assessment; hyperchloremic metabolic acidosis with a normal anion gap is present in all types of RTA

Urinalysis:
Analysis of urine biochemistry may also be useful in the evaluation of RTA; in patients with hyperchloremic metabolic acidosis and alkaline urine (i.e., pH > 5.5), RTA of some type should be strongly suspected
- Urine pH: Inappropriately, or persistently, alkaline (DIAGNOSTIC)

Acid load test:
Infusion of acid into blood with 100 mg/kg of ammonium chloride and check urine pH hourly and plasma HCO3 at 3-hour interval.
- A healthy person will be able to excrete acid and will decrease urine pH. Those with distal RTA cannot excrete acid and urine pH will remain basic despite increasingly acidic serum.
- Distal RTA: Measurement of the urine-to-blood (U-B) pCO2 gradient during an NaHCO3 infusion can be used to diagnose H+-ATPase secretory defect
- Proximal RTA: NaHCO3 loading test (during an IV NaHCO3, the increase in serum HCO3– above the reabsorption threshold will lead to fractional excretion of HCO3– > 15% or urine pH > 7.5 in patients with proximal RTA)
- Patients with proximal RTA should also be evaluated for Fanconi syndrome by assessment of serum and urine samples for glycosuria, hypophosphatemia, and hypouricemia.


Bicarbonate infusion test:
Fractional bicarbonate excretion is measured after an infusion of bicarbonate.
- Serum bicarbonate concentration approaches the normal level in the body after the infusion, which is more than the reabsorption threshold of the patient with type 2 Proximal RTA.
Urine Na test:
Type 4 RTA presents with persistently high urine Na despite restricted Na diet because of aldosterone deficiency or resistance.
Management
Type I & II RTA:
The basis of therapy is the continuous administration of appropriate amounts of alkali in the form of either bicarbonate or citrate. The amount of alkali administered should compensate for the urinary loss of HCO3− plus the amount of acid generated by the catabolism of proteins and the skeletal growth.
- Alkali therapy: Oral HCO3– (10-15 mEq/kg/day) + potassium supplementation
- Hydrochlorothiazide: Increases HCO3– tubular maximum (hypokalemia prevented with supplemental potassium)
- Fanconi syndrome: Phosphate supplementation
- Vitamin D supplementation
- Long-term follow-up (for renal stone complications)
Type IV RTA:
For hyperkalemic RTA, treatment and prognosis depends on the underlying cause. Potassium-retaining drugs should always be withdrawn. Hypoaldosteronism due to adrenal failure may respond adequately to replacement doses of fludrocortisone.
- Fludrocortisone therapy
- Loop diuretic (eg furosemide): Reduce risk of extracellular fluid volume expansion
- Alkali supplements (1.5 to 2 mmol/kg per 24 h)